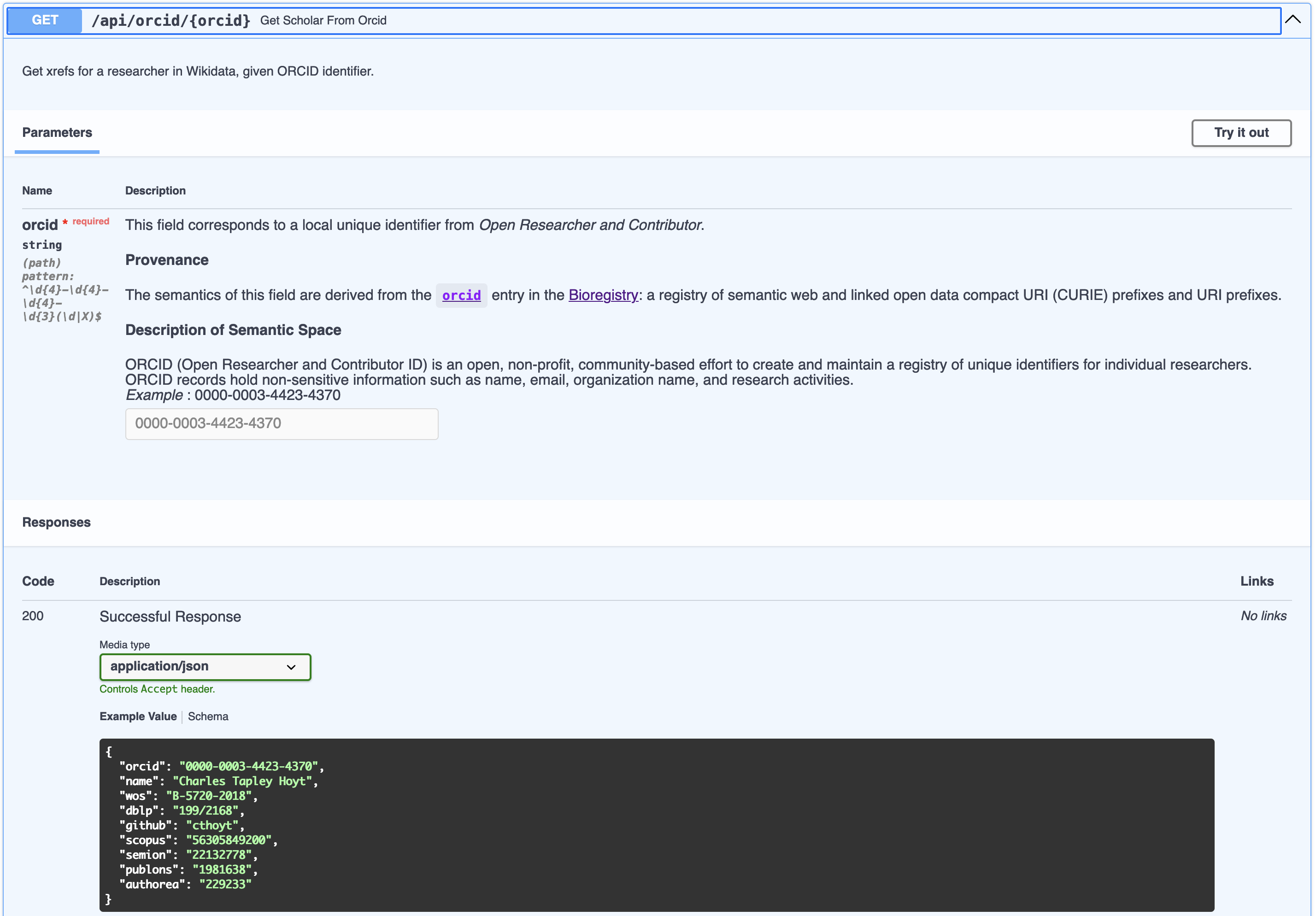
The Open Researcher and Contributor Identifier (ORCID) database is an invaluable resource that
supports the unambiguous identification of researchers. However,
its first party data dump is too complex, verbose, and unstandardized for
many use cases. This post describes open source software I wrote that
automates downloading, processing, and exporting ORCID into a more usable form. I
put the results
on Zenodo under the CC0 license.
Challenges to Overcome
ORCID currently contains on the scale of tens of million records, meaning that there isn’t a reasonable way to access
the data in bulk via its public API. As an alternative, ORCID dumps its public content once per year on FigShare. The
most recent (2023) dump is available at doi:10.23640/07243.24204912.
Previous versions are deposited with under different DOIs:
- 2022 ORCID Public Data File (https://doi.org/10.23640/07243.21220892.v4)
- 2021 ORCID Public Data File (https://doi.org/10.23640/07243.16750535.v1)
- 2020 ORCID Public Data File (https://doi.org/10.23640/07243.13066970)
- …
Unfortunately, this arrangement makes it difficult to automatically discover new versions without writing software that
searches FigShare programmatically and has a heuristic for guessing which might be a newer record. I don’t have a
solution for this yet, but I can imagine one.
Only making a yearly dump means that the derived artifacts can become out of date quickly. Other large
databases like PubChem make monthly and nightly dumps on their FTP servers which are deleted when no longer relevant.
For example, monthly dumps from more than a year ago can be safely deleted and nightly dumps only need to exist until
they are replaced by the next one. Since ORCID is using FigShare as an archival system, this would be a disk
space-intensive operation. Using the ORCID API or secondary data distribution via Wikidata could be good stopgaps
for consumers who want the most recent data.
ORCID distributes its data as XML. They also provide Java software
to convert it to JSON, but since 2018 are pretty adamant about not running this software and distributing the JSON
artifact themselves. This is unfortunate, since XML is hugely verbose both in terms of the way that data gets
structure and the markup itself. Further, the data structure contains a huge amount of provenance information that
isn’t useful for many downstream consumers (both in terms of when things were updated, by whom, and which API endpoint
could be used to update it further).
An example
from the JSON converter library also illustrates that converting from XML to JSON accomplish some obvious
simplifications that most users would want.
Another tricky thing about consuming the ORCID data is that the summary file that contains all the records is 32
gigabytes compressed and has a very strange internal file structure. This means that you either have to uncompress it,
which takes a long time with its tens of millions of files, or iterate through the file handles inside it. I also
haven’t figured out a good way to open a specific file inside the compressed archive beyond iterating through all the
handles. The file names themselves are also difficult to guess because of the directory structure used.
Solutions
I wrote a Python package, orcid_downloader that can automatically
download the right file from FigShare, iterate through the individual compressed XML files for each record, and process
them. The package can be used to iterate over records and process them in your own way like:
import orcid_downloader
for record in orcid_downloader.iter_records():
...
The main goal of this was also to create a simplified version of the ORCID data dump that is more
straightforwards, accessible, and smaller. I would imagine most people would be interested in just downloading the
results instead of rebuilding them, so the results of this process are posted on Zenodo at
doi:10.5281/zenodo.10137939. It uses Zenodo’s versioning system to
make sure that all different versions (both from updates to the yearly dump or improvements to the processing pipeline)
are all in the same Zenodo record.
So far, this includes the name, aliases, external identifiers, employers, education, and publications linked to PubMed.
Along the way, I realized that ORCID did not consistently ground educational institutions to the Research Organization
Registry (ROR) like it did for employers. I also had it double-check all groundings for employers,
since these were incomplete. I also did some minor string processing,
such as standardization of education types (e.g., Bachelor of Science, Master of Science), standardization of PubMed
references, and standardization of aliases (e.g., pruning off titles like Dr.)
The records.jsonl.gz file is a JSON Lines
file where each row represents a single ORCID record in a simple, well-defined
schema (see schema.json). Here are a few rows (
expanded for viewing comfort) as example:
{
"orcid": "0000-0001-5045-1000",
"name": "Patricio Sánchez Quinchuela",
"employments": [
{
"name": "Universidad de las Artes",
"start": 2021,
"role": "Especialista de Proyectos y docente",
"xrefs": {
"ror": "016drwn73"
}
},
{
"name": "Universidad Regional Amazónica IKIAM",
"start": 2019,
"end": 2021,
"role": "Director",
"xrefs": {
"ror": "05xedqd83"
}
}
],
"educations": [
{
"name": "Universidad Nacional de Educación a Distancia Facultad de Ciencias Políticas y Sociología",
"start": 2020,
"role": "Doctorando del Programa de Sociología",
"xrefs": {
"ringgold": "223339"
}
}
]
}
{
"orcid": "0000-0001-5101-6000",
"name": "Céline LEPERE"
}
{
"orcid": "0000-0001-5001-3000",
"name": "Vincent Nguyen",
"employments": [
{
"name": "Troy High School",
"start": 2020,
"role": "Student",
"xrefs": {
"ringgold": "289570"
}
}
]
}
{
"orcid": "0000-0001-5002-1000",
"name": "Sameer Abbood",
"employments": [
{
"name": "University of Al-Ameed",
"role": "Doctor of Philosophy"
}
]
}
Many records in ORCID are relatively unhelpful, i.e., ones that only have a name and no other (public) information.
Therefore, I created a high-quality subset that only contains records with at least one ROR-grounded employer, at least
one ROR-grounded education, or at least one publication indexed in PubMed. The point of this subset is to remove ORCID
records that are generally not possible to match up to any external information. It is listed in the same Zenodo record
as records_hq.jsonl.gz.
External Identifiers
An ORCID record has two places that make cross-references to external
nomenclature authorities:
- The “Websites & Social Links” section which allows a researcher to give a link with a name.
This is a trove of links to LinkedIn, Google Scholar, GitHub, and other external identifiers.
ORCID itself doesn’t standardize them, but using a combination of the Bioregistry and custom parsing,
many can be standardized.
- The “Other IDs” section is generated based on applications that connect to ORCID and send structured links. This
includes Scopus, Web of Science (formerly ResearcherID), Loop, and some others. This also needs quite a bit
of standardization, probably due to a combination of bugs in ORCID, bugs in external services, legacy data, and other
things.
Reflections
I found several interesting things while parsing these sections:
- I discovered several new nomenclature authorities that weren’t already registered in
the Bioregistry. This includes Loop, Digital Author ID (Dutch),
Authenticus (Portuguese), Dialnet (Spanish), SciProfiles, Ciência (Portuguese), and KAKEN (Japanese). I still have to
send new prefix requests for these.
- I found several fields that were totally junk or didn’t make sense. For example, there is sometimes a reference to
the ORCID record itself. There are also references to external IDs that aren’t really IDs, or at least don’t follow
enough of the guidelines
from Identifiers in the 21st Century
to be useful.
- Both the “Websites & Social Links” and “Other IDs” needed standardization. In some places, this was as easy as
using the Bioregistry prefix standardization, but in other places required more custom URL parsing. This is
especially true for Google Scholar, which can appear with a number of domain names (e.g., https://scholar.google.com
or https://scholar.google.es). The local unique identifier appears here inside the URL parameters, which can be in
any order along with the language tag, so this needs URL parsing instead of more simple URI prefix handling a la
the
curies Python Package.
Summary
Here’s a breakdown of the top external cross-references, standardized with the Bioregistry (where possible). Note that
this was prioritized by the most common
cross-references, and is not complete. To capture all would be a lot of work and require many more corner cases
to less common services. I also threw away links to non-professional social networks like Facebook/Instagram.
I also made the value judgement to throw away links to Twitter since it doesn’t reflect open and inclusive scientific
community values anymore.
| Resource |
Count |
scopus |
1,400,735 |
wos.researcher |
608,543 |
sciprofiles |
259,654 |
loop |
229,224 |
linkedin |
191,321 |
researchgate |
125,242 |
google.scholar |
52,131 |
github |
13,397 |
gnd |
7,621 |
isni |
4,105 |
dai |
1,982 |
authenticus |
1,422 |
dialnet |
1,210 |
wikidata |
147 |
The results of this process are first available as part of the records file(s). Second, they are available through
a dedicated file (sssom.tsv.gz) in
the Simple Standard for Sharing Ontological Mappings (SSSOM)
that solely focuses on the cross-references. Here’s what the first few lines of that file look like:
What are Cross-References Useful For?
There are many different nomenclature authorities because each have different goals and data models associated with
records. Different communities also value different nomenclature authorities differently. For example, life scientists
are more often using ORCID when publishing, but including persistent identifiers in publications is not yet common
in computer science papers. Further, computer scientists more often link to
their DBLP,
arXiv, OpenReview, or other computer-science focused pages.
When assembling data and knowledge from more than a single resource, it’s important to resolve the identifiers
used for researchers to a single identifier - it’s not good if different knowledge is connected to an ORCID and a
DBLP for a single researcher. This can be resolved using a combination of semantic mappings (i.e., cross-references)
and software for the large-scale automated assembly of mappings such as the
Semantic Mapping Reasoning Assembler (SeMRA).
It allows for specifying a priority list of nomenclature authorities to assemble coherent knowledge from ORCID
and other sources simultaneously.
This is also a much more valuable process when combining other mapping resources. Wikidata has put a lot of effort
into capturing bibliographic metadata, especially to support the Scholia project.
The following SPARQL query against Wikidata shows that there are 1,811,573 (about 10% of all ORCID records) links from
Wikidata entries to ORCID as of June 2024. Run it yourself.
SELECT (count(DISTINCT ?orcid) as ?total)
WHERE { ?item wdt:P496 ?orcid }
OpenCheck tried to create mappings between Twitter, GitHub, and ORCID using their APIs, but
became defunct when Twitter shut off their public APIs.
OpenAlex, Microsoft Academic Graph,
Open Academic Graph, and other
bibliographic knowledge graphs all have to address this problem internally, as well.
Other mapping resources probably exist, please let me know if you’re aware of other ones!
Affiliations
ORCID breaks up affiliations into several blocks: educations, employments, invited positions, etc.
I’m focusing on educations and employments here. The processing code could be extended for the others later.
Employments contained ROR references but educations didn’t, and even when they’re available, they’re incomplete.
PyOBO has implemented a loader for ROR. Any resource loaded through PyOBO
can also be used for named entity normalization through an interface to Gilda.
This allowed for grounding of a large number of missing education and employer entries. Here’s the total number of
cross-references made from educations and employments to ROR and other nomenclature authorities.
Education Roles
Both educations and employments also have associated roles. These do not use a controlled vocabulary, but there
are a number of patterns that could be standardized. This task had to be split between education and employment entries.
I just focused on education entries for now. The “role” field in each education entry corresponds to the degree.
I started by looking for existing resources (both structured and unstructured) that have lists of degrees. Here
are a few things I found:
- https://degree.studentnews.eu lists degrees conferred in the EU/Europe
- https://github.com/vivo-ontologies/academic-degree-ontology is an incomplete/abandoned
effort from 2020 to ontologize degree names
-
Wikidata has a class for academic degree https://www.wikidata.org/wiki/Q189533. Its
SPARQL query service <https://query.wikidata.org>_ can be queried with the following,
though note that the Wikidata class hierarchy is broken in several places.
SELECT ?item ?itemLabel WHERE {
?item wdt:P279* wd:Q189533 .
SERVICE wikibase:label { bd:serviceParam wikibase:language "[AUTO_LANGUAGE],en". }
}
In the end, these were either incomplete or not organized well enough to directly use. It also turns out
that people often conflate the degree (e.g., Master of Science) with the field that it’s in (e.g., Chemistry) or
include some combination (e.g., Master of Science in Chemistry). This meant that a lot of string processing and
heuristics would be required on top of lexical approaches. Instead, I took the tried-and-true method of listing
the strings by frequency and just curating from the top. The results are in
https://github.com/cthoyt/orcid_downloader/blob/851af81d8aacebf2768bfc591080cbceab2047cc/src/orcid_downloader/standardize.py.
Of course, this is incomplete in many ways, and could be extended to capture further cases. I also found that there
are a huge number of Spanish and Portuguese entries that I needed help from my international friends to get the best
translations (since the meaning is pretty subtle for many). Further, the results would be more useful as a proper
ontology that could extend and replace VIVO’s Academic Degree Ontology. I’ll leave this for future work. Here’s
a summary of the most frequent roles that have been standardized so far:
| Education Role |
Count |
| Doctor of Philosophy |
1,231,845 |
| Master |
426,625 |
| Bachelor |
402,297 |
| Master of Science |
360,052 |
| Bachelor of Science |
322,853 |
| Doctor of Medicine |
202,884 |
| Bachelor of Arts |
108,778 |
| Master of Arts |
78,601 |
| Postdoctoral Researcher |
37,548 |
| Bachelor of Medicine, Bachelor of Surgery |
27,722 |
| Master of Technology |
22,473 |
| Bachelor of Technology |
21,465 |
| Bachelor of Engineering |
21,345 |
| Diploma |
20,695 |
| Master of Business Administration |
19,660 |
| Master of Philosophy |
18,069 |
| Master of Education |
18,055 |
| Master of Public Health |
17,760 |
| Bachelor of Education |
17,376 |
| Master of Engineering |
14,260 |
Authorship
Authorships are extracted and standardized in
the pubmeds.tsv.gz file, which contains an
ORCID column and PubMed column that has been pre-sanitized to only contain local unique identifiers. This information is
also available through the main records file.
While the field inside the XML data is supposed to contain local unique identifiers, there was a huge variety of what
actually showed up there. This included local unique identifiers (i.e., 36402838), local unique identifiers with junk
attached (e.g., 36402838/), valid Compact URIs (CURIEs), invalid CURIEs, URIs, free text that’s totally irrelevant.
Overall, there were 3,175,196 (99.85%) that were valid local unique identifiers, 2,832 (0.09%) that were able to be
cleaned up, and 2,080 (0.07%) that were junk and couldn’t be salvaged. Inside the junk were a few things:
- DOIs
- PMC identifiers,
- a few stray strings that contain a combination of pubmed, PMC, and DOIs
- a lot with random text (keywords)
- some with full text citations
Later, other identifier types could be added in here too.
Lexical Indexing
One of the original goals of processing ORCID in bulk was to ground and disambiguate author lists in publications.
Therefore, I made two pre-built Gilda indexes for named entity recognition (NER) and named entity normalization (NEN).
One contains all records, and the second is filtered to high-quality records. The following Python code snippet can be
used for grounding:
from gilda import Grounder
url = "..."
grounder = Grounder(url)
results = grounder.ground("Charles Tapley Hoyt")
The ORCID downloader also has its own extension that does a smarter job with caching and some clever name preprocessing
import orcid_downloader
results = orcid_downloader.ground_researcher("Charles Hoyt")
Ontology Artifact
The file orcid.ttl.gz is an OWL-ready RDF file
that can be opened in Protégé or used with
the Ontology Development Kit.
It can also be converted into OWL XML, OWL Functional Notation, or other OWL formats
using ROBOT. This artifact can serve as a replacement for the ones generated
by https://github.com/cthoyt/orcidio,
which was a smaller-scale way of turning ORCID records for contributors to OBO Foundry
ontologies into a small OWL file. Now, the export here contains all ORCID records with names.
@prefix orcid: <https://orcid.org/> .
@prefix rdfs: <http://www.w3.org/2000/01/rdf-schema#> .
@prefix oboInOwl: <http://www.geneontology.org/formats/oboInOwl#> .
@prefix owl: <http://www.w3.org/2002/07/owl#> .
@prefix skos: <http://www.w3.org/2004/02/skos/core#> .
@prefix human: <http://purl.obolibrary.org/obo/NCBITaxon_9606> .
human: a owl:Class ;
rdfs:label "Homo sapiens"^^xsd:string .
orcid:0000-0001-5000-5000 a human: ;
rdfs:label: "Joel Adam Gordon"^^xsd:string ;
oboInOwl:hasExactSynonym "Joel Gordon"^^xsd:string .
orcid:0000-0001-5099-6000 a human: ;
rdfs:label: "Debashis Bhowmick"^^xsd:string ;
oboInOwl:hasExactSynonym "Bhowmick D. S."^^xsd:string ;
oboInOwl:hasExactSynonym "D. S. Bhowmick"^^xsd:string ;
oboInOwl:hasExactSynonym "Debashis S Bhowmick"^^xsd:string ;
skos:exactMatch "scopus:57214299968" .
orcid:0000-0001-5084-9000 a human: ;
rdfs:label: "Luana Licata"^^xsd:string ;
skos:exactMatch "loop:1172627"^^xsd:string ;
skos:exactMatch "scopus:6603618518"^^xsd:string .
It’s still TBD on the best way to encode the cross-references.
Code
The artifacts described here were all automatically generated with code in https://github.com/cthoyt/orcid_downloader.
Like I mentioned a few times throughout, this is a work in progress. Doing practical data science is hard work, and
there is a lot of room for improvement. I’m still recovering from burnout, so working at a slow pace only when I
felt inspired also was fine for me. I know there are lots of things I would like to improve given more motivation,
but that will have to wait for now.
]]>